You are here
Om kliniske studier
Denne side indeholder forståelige og pålidelige oplysninger om kliniske studier. Klik på nedenstående spørgsmål for klare og præcise svar.
Hvorfor er kliniske studier vigtige?
De, der deltager i et klinisk forsøg, enten som patienter eller som sunde frivillige, bidrage til fremme af medicin og en bedre fremtid for patienterne.
Eksisterende medikamenter (lægemidler/stoffer) kan enten være uegnede eller ikke effektive for mange patienter. Derfor gennemfører hundreder af tusindvis af forskere verden over forskning om nye lægemidler og behandlinger. Dog kan disse stoffer ikke markedsføres uden først at gennemføre kliniske studier med raske frivillige og patienter. Disse kliniske undersøgelser er udført for at finde ud af den langsigtede sikkerhed, effektivitet og effekt af medicinen. Derudover sammenlignes nye lægemidler eller behandlinger med eksisterende.
Det er nødvendigt, at nok mennesker deltager i disse undersøgelser, så videnskabeligt underbyggede konklusioner kan drages.
Hvad er klinisk forskning?
Når en ny medicin udvikles, skal det bevises, at denne nye medicin er sikker, og at den virker, før det kan markedsføres.
Udvikling af medicin er opdelt i præklinisk forskning inden for testrør og dyr på den ene side og klinisk forskning med mennesker på den anden side. Først efter at det er blevet bevist, at det nye produkt er sikkert og effektivt i dyreforsøg, kan det undersøges hos mennesker.
De tests, der gennemføres hos mennesker for at finde ud af, om det nye produkt er sikkert og effektivt kaldes kliniske studier. Disse studier udføres i et sæt af fire på hinanden følgende faser (I, II, III og IV). Målet med et klinisk studie afhænger af det nye produkt, der undersøges.
Hvad er de faser af klinisk forskning hos mennesker?
- Prækliniske forskning inden for testrør og dyr
- Klinisk forskning med mennesker
Klinisk forskning består af strengt kontrollerede kliniske studier i 4 på hinanden følgende faser.
- Fase 1 studier - Er stoffet sikkert?
Det potentielle nye lægemiddel administreres normalt til en lille gruppe (20-100) af sunde frivillige til at studere de generelle virkninger (distribution, metabolisme og eliminering samt eventuelle bivirkninger) af det nye lægemiddel. - Fase 2 studier – Virker stoffet?
Hvis resultaterne fra fase I-studie viser, at det potentielle nye lægemiddel er sikkert, startes en fase II studie i en lille gruppe udvalgte patienter (100 til 500), der deltager på frivillig basis. I løbet af denne fase undersøger forskerne om, hvorvidt det nye lægemiddel virker for den sygdom, som den blev udviklet til. Patienterne, der kan deltage, vælges ud fra strenge kriterier for integration og udelukkelse. Dette gøres for at minimere variationen i gruppen, således at der kan laves videnskabeligt underbyggede konklusioner. - Fase 3 studier - Virker stoffet bedre, lige så godt eller mindre godt end eksisterende lægemidler eller behandlinger?
Det potentielle nye stoffer undersøges i en større gruppe patienter (1000 til 5000) for at studere dens langsigtede effektivitet og sikkerhed. I denne fase er lægemidlet sammenlignet med eksisterende lægemidler eller behandlinger. Hvis det er bevist, at stoffet giver tilstrækkelige eller bedre resultater og er sikkert, får det officielt markedsføringstilladelse. Fra det øjeblik, er det nye lægemiddel tilgængelig på markedet og kan ordineres af læger. - Fase 4 studier - Hvad er de langsigtede effekter?
Det nye lægemiddels sikkerhed fortsætter med at blive overvåget, selv efter at den er blevet lanceret på markedet. I fase IV studier, ser forskere primært på mulige bivirkninger, der forekommer sporadisk og på langsigtede komplikationer. Desuden undersøger forskere undertiden om stoffet er effektivt til andre sygdomme eller lidelser.
Hvem gennemfører kliniske studier?
Der er en differentiering mellem (i) kommercielle studier og (ii) ikke-kommercielle eller akademiske studier. Initiativtager til en kommerciel undersøgelse er et farmaceutisk selskab eller en virksomhed, der fremstiller medicinsk udstyr. Med et akademisk studie, initiativtager er en efterforsker fra et hospital.
Begge typer af studier skal følge de samme strenge regler. I begge tilfælde er et studieemne lige så godt beskyttet og har samme rettigheder og forpligtelser.
I kommercielle studier er der ingen direkte kontakt mellem patienter og initiativtager. Al kontakt går igennem en deltagende studieplads og en efterforsker, der er forbundet med studiet. Undersøgeren sikrer, at forsøgspersoner overvåges, følges op og undersøgt.
Hvem kan deltage i et klinisk studie?
Både raske frivillige og patienter kan være involveret i et studie.
Hvert studie har specifikke betingelser, og disse er altid beskrevet i forsøgsprotokollen. En klinisk studieprotokol indebærer alle af undersøgelsens regler og procedurer. Dette er referencebogen eller playbook, som det var, for korrekt gennemførelse af et studie. Protokollen skal altid godkendes af en medicinsk etisk komité og af de respektive myndigheder, før studiet kan begynde.
Betingelserne for deltagelse kaldes også inklusions- og eksklusionskriterier.
Mulige deltagere:
- skal opfylde studiebetingede forhold;
- er og skal grundigt informeres ud fra oplysningsformularen;
- skal forstå, hvad deltagelse indebærer, herunder hvad de mulige risici og fordele er;
- skal kunne deltage helt frivilligt, som forklaret i oplysningsformularen;
- skal være forsikret mod mulig skade;
- skal kunne stoppe deltagelsen i undersøgelsen til enhver tid;
- skal godkende og underskrive et dokument, kendt som patientoplysning og informeret samtykkeformular ved studiestartens begyndelse.
Et det sikkert at deltage i et klinisk studie? Hvem garanterer denne sikkerhed?
Kliniske studier er meget strengt reguleret for at holde risikoen for forsøgspersonerne til et minimum. For at beskytte studiematerialers rettigheder, integritet og fortrolighed samt undersøgelsesdataets troværdighed er de internationale etiske og videnskabelige forhold angivet i Helsinki-erklæringen og retningslinjen for god klinisk praksis (GCP).
Før et klinisk studie kan startes, skal først indhentes godkendelse til det fra et anerkendt medicinsk etik komité samt fra regeringen. Begge de ovennævnte enheder giver anbefalinger og vurderer, om det nye lægemiddel er sikkert og, om fordelene ved det nye lægemiddel opvejer de potentielle risici, forbundet med studiet. Det er først efter en positiv udtalelse (= godkendelse) modtages, at studiet kan startes.
Den kompetente lægelige etiske komité overvåger deltagernes interesser i hele studiens varighed. Studiet kan stoppes på ethvert tidspunkt, for eksempel på grund af uacceptable bivirkninger.
Hvad er de mulige risici ved at deltage i et klinisk studie?
Først udarbejdes en studieprotokol for at gennemføre en klinisk undersøgelse. En klinisk studieprotokol indebærer alle af undersøgelsens regler. Dette er den referencebogen eller playbook, som det var, for korrekt gennemførelse af et studie. Protokollen skal altid godkendes af en medicinsk etisk komité og af de respektive myndigheder, før studiet kan begynde. Den medicinske etiske komité ure også over deltagernes interesser under studiet.
På trods heraf, kan der være risici eller ulemper forbundet med at deltage i et klinisk studie. En undersøgelse risikoprofil analyseres af forskellige parter (virksomheden, sponsorerer studiet, læger, etiske udvalg og regeringen), og studiet kan kun startes, hvis fordelene opvejer de mulige risici.
Risici / ulemper kan omfatte følgende:
- Stoffet eller behandlingen er normalt nyt og undersøges stadig. Det betyder, at meget ofte ikke alle de bivirkningerne af lægemidlet er kendt. Risikoen for ukendte bivirkninger varierer per studie og kan ikke altid helt forudsiges. Det er derfor, at efterforskeren giver emnet oplysninger om dette på forhånd. Selvfølgelig overvåges alle bivirkninger nøje af både undersøgeren og de etiske komitéer, der overvåger alle deltageres sikkerheden. Desuden kan en deltager beslutte at stoppe deltagelsen i studiet på ethvert tidspunkt.
- Det er muligt, at det nye lægemiddel kan have nogen merværdi.
- I fase II og fase III kliniske studier ved deltageren normalt ikke, om han / hun modtager stoffet eller ej. Hvis det er etisk, kan forsøgsprotokollen angive en kontrolgruppe, der ikke vil modtage det aktive produkt, men en placebo i stedet.
- Deltagelse kan kræve en ekstra indsats fra undersøgelsen med forbehold på grund af ting såsom ekstra hospitalsbesøg, eksamener eller procedurer (såsom udtagning af blodprøver).
Spørgsmål om din eventuelle deltagelse
Overvejer du at deltage i et klinisk studie? Klik på nedenstående spørgsmål om din mulige deltagelse.
Hvorfor skal du deltage i et klinisk studie, og hvad er de mulige fordele ved at deltage?
- Ved at deltage i et klinisk studie, støtter du medicinsk forskning, og derved bidrage til udviklingen af medicinsk videnskaben.
- Ved at deltage som patient du kan drage fordel af den seneste sundhedspleje samt en mulig ny behandling for din sygdom, hvis virkning og sikkerhed undersøges.
- Resultaterne af studierne kan også hjælpe fremtidige patienter ved at udvikle et potentielt nyt lægemiddel, en ny vaccine, en bedre diagnostisk test, …
- Forskere kan udføre flere tests end hvad han / hun normalt ville gøre for at overvåge virkningerne af det administreret lægemiddel så tæt som muligt. Du vil ikke blive opkrævet for disse yderligere behandlinger og / eller prøver i løbet af studiet.
- Undersøgelserne er strengt fortrolige og dit privatliv vil ikke blive bragt i fare på noget tidspunkt.
Der er dog ingen garanti for, at din deltagelse vil gavne dig personligt. Du skal også tage hensyn til den ekstra indsats og risici, der kan være forbundet med et studie. De mulige fordele, men også de ulemper og mulige risici ved et specifik studie, vil altid blive forklaret og diskuteret med dig på forhånd.
Hvem beslutter, om du kan deltage i et klinisk studie?
Forskeren vurderer, om du kan deltage i et klinisk forsøg.
Deltagelse i et klinisk studie er altid helt frivilligt. Hvis du beslutter dig ikke at deltage i et studie, vil dette på ingen måde påvirke din videre behandling og dit forhold til din behandlende læge. Du kan også beslutte at stoppe deltage i studiet på ethvert tidspunkt.
Du kan finde flere detaljer om vores rolle i vores henvisningspolitik.
Hvad sker der med dine data under et klinisk studie?
Hvis du opfylder udvælgelseskriterierne og beslutter at deltage i et klinisk studie, vil din deltagelse og alle dine personlige og medicinske data behandles fortroligt i overensstemmelse med lokal lovgivning. Under et klinisk studie vil dine data blive krypteret, når indsamles og håndteres. Disse data vil kun blive behandlet og analyseret i en videnskabelig sammenhæng, der er relateret til studiet. I senere videnskabelige publikationer af studier, vil der ingen oplysninger blive delt som personligt kunne identificere undersøgelsens deltagere.
På ingen tid vil din forsker dele med C-Lys de medicinske data, der indsamles i løbet af et klinisk forsøg,. C-Lys er kun involveret i den første udvælgelse af kandidatemner.
Hvad sker der, hvis du lider skade under et studie?
Hvert klinisk studie er omhyggeligt designet for at holde risici til et minimum. Dog kan uforudsete problemer ikke udelukkes. Det er derfor, at loven siger, at sponsor er forpligtet til at indgå forsikring for hvert klinisk studie for at beskytte deltagerne. C-Lys er kun involveret i den første udvælgelse af forsøgskandidater og er ikke ansvarlig for din deltagelse i studiet. For mere oplysning om forsikringen, kan du kontakte undersøgelsens efterforsker.
Hvis du er interesseret i at deltage i et klinisk studie, kan efterforskeren forklare undersøgelsens mål og de mulige fordele for dig på forhånd. Din læge vil også påpege, at sådanne fordele ikke kan garanteres. Bivirkninger kan altid forekomme på grund af den forskningsfase, hvor lægemidlet eller behandling i øjeblikket befinder sig i.
Vil du modtage økonomisk kompensation for at deltage i et klinisk studie?
Raske frivillige, der deltager i fase I studier normalt modtage økonomisk kompensation, der beregnes ud fra den belastning, der er forårsaget af testene (antal blodprøver, der skal tages, antal overnatninger, hospitalsophold …), som du vil gennemgå for undersøgelsen. Økonomisk kompensation er normalt ikke juridisk tilladt for deltagelse i fase II, III og IV studier som patient. I nogle tilfælde kan du være berettiget til refusion af dine parkering og rejseudgifter. I begge tilfælde kan forsker i undersøgelsen give dig mere oplysning om dette.
Økonomisk kompensation eller refusion af udgifter betales altid af studiens sponsor. I betragtning af, at C-Lys kun er involveret i den første udvælgelse af forsøgspersoner, er C-Lys ikke ansvarlig for at betale erstatning til emner i alle tilfælde.
Hvor skal du rapportere mulige bivirkninger?
Når du deltager i et klinisk studie, er det ekstremt vigtigt at rapportere eventuelle bivirkninger eller bivirkninger til din behandlende læge. Dette hjælpe at sikre sikkerheden af lægemidlet eller behandlingen.
Denne C-Lys hjemmeside er ikke beregnet til at rapportere mulige bivirkninger. til kliniske studier, gælder en streng regulering for at beskytte deltagerne. På grund af denne unikke juridiske situation, kan C-Lys medarbejdere men må ikke besvare spørgsmål om medicinske produkter, medicinske behandlinger, bivirkninger, eller specifikke kliniske studier. Vi har kun de oplysninger, du kan finde på denne hjemmeside. Oplysninger om specifikke studier er udarbejdet i samråd med forskerne og godkendt af en kompetent etisk komité.
Denne C-Lys hjemmeside er ikke beregnet til at dele din erfaring inden for et klinisk studie. Det kan være fristende at tale med andre, der er ligesom du at deltage i et bestemt klinisk studie. Du kan begynde at fortælle om de mulige bivirkninger eller om dine oplevelse, men det er vigtigt ikke at gøre dette. Det kan trods alt påvirke resultatet af studiet. Der er faktisk noget som forslagets kraft: høring om en bivirkning kan være nok til faktisk at få denne bivirkning.
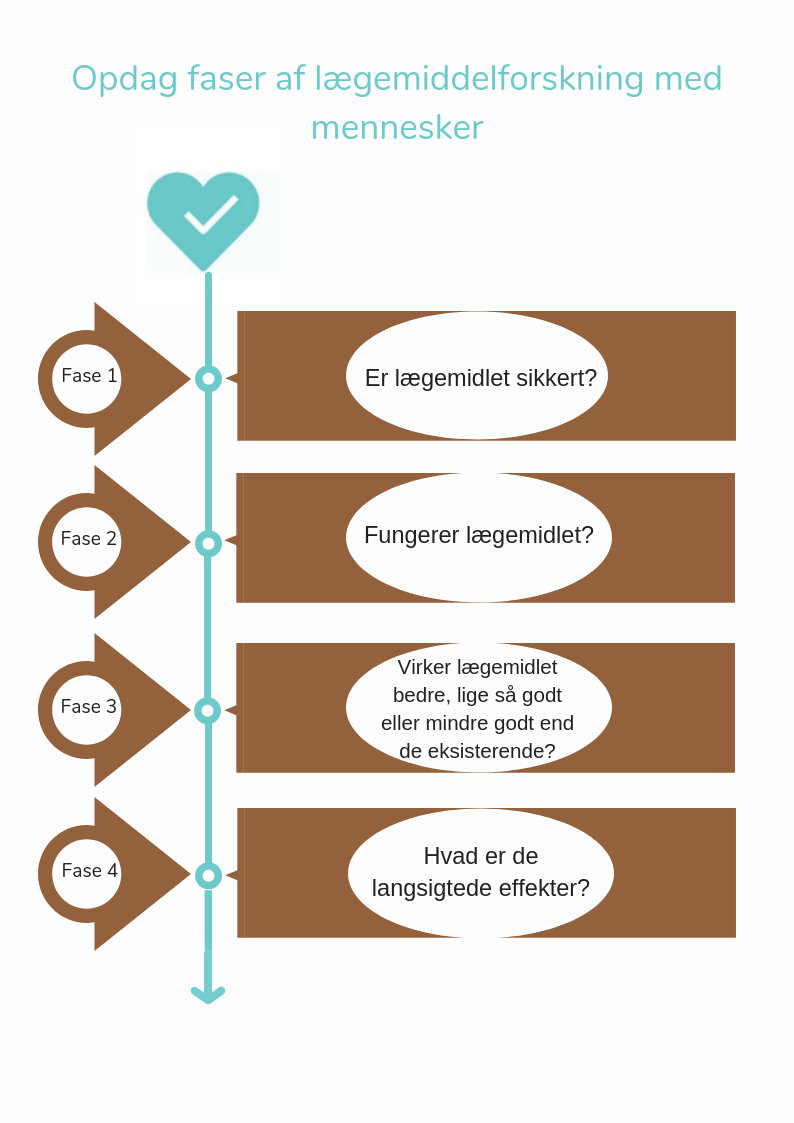
Kliniske studier: fra fase 1 til fase 4.